
Une protéine de la bactérie Staphylococcus aureus
Fin novembre, l'équipe DeepMind de Google a annoncé que son système d'apprentissage en profondeur AlphaFold avait atteint des niveaux de précision sans précédent pour résoudre le problème du repliement des protéines , un problème difficile en biochimie informatique.
Quel est le problème et pourquoi est-il si difficile à résoudre?
Les protéines sont de longues chaînes d'acides aminés. Votre ADN code pour ces séquences et l'ARN aide à fabriquer des protéines selon ce plan génétique. Les protéines sont synthétisées sous forme de chaînes linéaires, mais se replient ensuite en structures sphériques complexes (voir la photo au début de l'article).
Une partie de la chaîne peut s'enrouler en une spirale serrée. " α-hélice . "L'autre partie peut se plier d'avant en arrière pour former une figure large et plate," feuille β ":
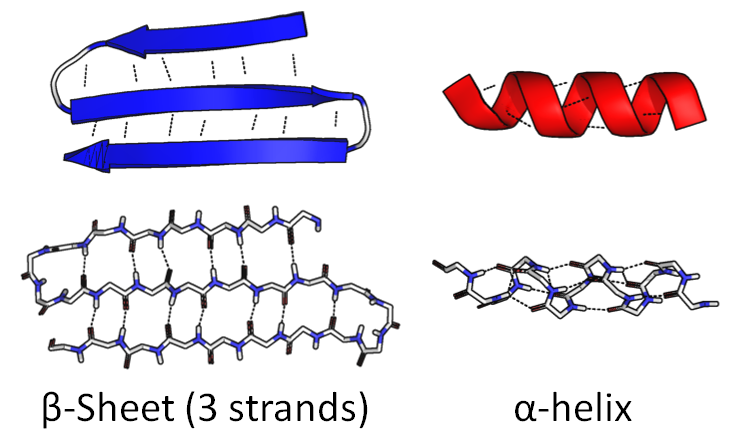
La séquence d'acides aminés elle-même est appelée la structure primaire . Ces figures sont appelées structure secondaire .
Ces composants eux-mêmes se plient également pour former des formes complexes uniques. C'est ce qu'on appelle la structure tertiaire :
une enzyme tirée de la bactérie Colwellia psychrerythraea RRM3

protéine
semble désordonnée. Pourquoi cette boule d'acides aminés enchevêtrée est-elle si importante?
La structure des protéines n'est pas aléatoire! Chaque protéine se replie dans une structure distincte, unique et largement prévisible, ce qui est essentiel pour son bon fonctionnement. En raison de sa forme physique, la protéine est bien adaptée aux structures avec lesquelles elle peut se lier. D'autres propriétés physiques sont également importantes, en particulier la distribution de la charge électrique sur la protéine. Sur la photo, une charge positive est indiquée en bleu, une charge négative en rouge:

Répartition de la charge de surface sur la protéine porteuse lipidique des plantes 1 de riz
Si une protéine est essentiellement une nanomachine auto-assemblante, alors le but principal d'une séquence d'acides aminés sera de produire sa forme unique, sa distribution de charge et tout ce qui détermine la fonction de la protéine. Comment exactement ce processus se déroule n'est pas encore tout à fait clair - c'est aujourd'hui un domaine de recherche actif.
Dans tous les cas, la compréhension de la structure est importante pour comprendre son fonctionnement. Cependant, la séquence d'ADN ne définit que la structure primaire de la protéine. Comment connaissons-nous ses structures secondaires et tertiaires, c'est-à-dire la forme exacte que prendra cet enchevêtrement?
Ce problème s'appelle le problème du repliement des protéines et il existe deux approches de base: la mesure et la prédiction.
Les méthodes expérimentales peuvent mesurer la structure d'une protéine. Cependant, ce n'est pas si facile à faire: les structures ne sont pas visibles au microscope optique. Pendant longtemps, la cristallographie aux rayons X a été la principale méthode d'étude des structures. En plus de cela, la résonance magnétique nucléaire a été utilisée, et récemment une nouvelle technologie est apparue, la microscopie cryoélectronique .

Diagramme de diffraction des rayons X de la protéase du SRAS
Cependant, ces méthodes sont coûteuses, complexes et prennent du temps, et de plus, elles ne fonctionnent pas avec toutes les protéines. En particulier, les protéines intégrées dans la membrane cellulaire - le même récepteur de l' enzyme de conversion de l'angiotensine 2 (ACE2) auquel le virus COVID-19 se lie - se replient dans la bicouche lipidique cellules, et il est très difficile de cristalliser.

La structure de la membrane cellulaire
Par conséquent, nous avons pu démonter la structure d'un infime pourcentage des protéines séquencées . La base de données universelle des protéines contient 180 millions de séquences, tandis que la base de données des structures de protéines tridimensionnelles ne contient que 170 000 positions.
Nous avons besoin d'une meilleure méthode.
* * *
Rappelons que les structures secondaires et tertiaires des protéines sont essentiellement fonction de la structure primaire que nous connaissons par séquençage. Et si, au lieu de mesurer la structure d'une protéine, on pouvait la prédire?
C'est la tâche de prédire la structure des protéines. Les biochimistes informatiques y travaillent depuis des décennies.
Comment pouvez-vous l'aborder?
La manière la plus évidente est de simuler directement la physique du processus. Nous simulons les forces pour chaque atome, en tenant compte de son emplacement, de sa charge et de ses liaisons chimiques. Nous comptons les accélérations et les vitesses, et faisons défiler étape par étape l'évolution du système. C'est ce qu'on appelle la «dynamique moléculaire».

Supercalculateur " Anton " par DE Shaw Recherche

Supercalculateur IBM Blue Gene
Puzzle en ligne Foldit
Le problème est que cette approche est extrêmement gourmande en calculs. Une protéine typique contient des centaines d'acides aminés, c'est-à-dire des milliers d'atomes. L'environnement compte également: lors du pliage, la protéine interagit avec l'eau environnante. Par conséquent, il est nécessaire de simuler le comportement d'environ 30 mille atomes. Dans ce cas, une interaction électrostatique se produit entre chaque paire d'atomes, c'est-à-dire qu'avec une estimation approximative, nous obtenons 450 millions de paires, un problème de complexité O (N2). Il existe des algorithmes intelligents qui réduisent sa complexité à O (N log N). De plus, pour la simulation, il est nécessaire de calculer 10 9 -10 12 pas. Mal de tête exceptionnel.
D'accord, mais nous n'avons pas besoin de simuler tout le processus de pliage. Une autre approche suggère de trouver une structure avec une énergie potentielle minimale. Habituellement, les objets ont tendance à se reposer avec le moins d'énergie, cette approche heuristique est donc justifiée. L'énergie peut être calculée par le même modèle de dynamique moléculaire, ce qui nous donne l'ampleur des interactions. Avec cette approche, nous pouvons essayer un tas de candidats et choisir la structure avec le moins d'énergie. Le problème, bien sûr, est de savoir d'où proviennent les structures. Il y en a tout simplement trop - le biologiste moléculaire Cyrus Levintol a calculé qu'il pourrait y en avoir environ 10 300 . Naturellement, vous pouvez utiliser une approche plus intelligente que la force brute aléatoire. Mais il y en a encore trop.
Par conséquent, de nombreuses tentatives ont déjà été faites pour accélérer ces calculs. Anton, un supercalculateur de DE Shaw Research, utilise un matériel spécial - des circuits intégrés spéciaux. IBM utilise également le supercalculateur bio Blue Gene. Stanford a lancé le projet Folding @ Home, en utilisant la puissance distribuée des ordinateurs personnels. Le projet Foldit de UW a transformé le pliage en un jeu pour ajouter l'intuition humaine au calcul.
Pourtant, pendant longtemps, aucune technologie n'a été en mesure de prédire une large gamme de structures protéiques avec une grande précision. Lors du concours CASP organisé deux fois par an, où les résultats des algorithmes sont comparés aux structures mesurées expérimentalement, les premières places ont reçu des prédictions avec une précision de 30 à 40%. Jusque récemment:

Meilleure précision prédictive médiane d'équipe dans la catégorie de modélisation gratuite
Comment fonctionne AlphaFold? Il utilise plusieurs réseaux de neurones profonds pour apprendre différentes fonctions associées à chaque protéine. L'une des fonctions clés est de prédire les distances résultantes entre les paires d'acides aminés. Cela amène l'algorithme à la structure finale. Dans une variante de l'algorithme (décrite dans les revues Nature and Proteins ), la fonction potentielle de cette prédiction a été dérivée, à laquelle la descente de gradient la plus simple a été appliquée, ce qui a étonnamment bien fonctionné.
Le principal avantage d'AlphaFold par rapport aux méthodes précédentes est qu'il n'a pas besoin de faire d'hypothèses sur les structures. Certaines méthodes fonctionnent en décomposant les protéines en sections, en comptant chacune d'elles, puis en remettant tout en place. AlphaFold n'en a pas besoin.
Apparemment, DeepMind considère le problème de pliage résolu, ce qui me semble une simplification excessive, mais dans tous les cas, leur progrès est significatif. Les experts non affiliés à Google utilisent des épithètes telles que « fantastique » et « révolutionnaire ».
Le génie génétique dispose désormais de deux outils puissants, CRISPR et le repliement des protéines. Peut-être que les années 2020 seront pour la biotechnologie ce que les années 70 étaient pour l'informatique.
Félicitations aux chercheurs de DeepMind pour cette percée!