
Dans un article récemment publié dans Physical Review Research, nous démontrons comment l'apprentissage profond simplifie la solution d'équations de mécanique quantique fondamentales pour des systèmes réels. Dans le même temps, non seulement une question scientifique fondamentale est résolue, mais aussi des perspectives d'utilisation pratique des résultats obtenus dans le futur s'ouvrent.
Les chercheurs pourront prototyper de nouveaux matériaux et composés in silico avant d'essayer de les synthétiser en laboratoire. A également publié le codede cette étude; ainsi, les équipes de physique et de chimie computationnelles peuvent s'appuyer sur leurs travaux et les appliquer à une variété de problèmes. Dans le cadre de l'étude, une nouvelle architecture de réseau de neurones, Fermionic Neural Network ou FermiNet, a été développée, qui est bien adaptée pour simuler l'état quantique de grandes collections d'électrons - et toutes les liaisons chimiques sont basées sur des électrons. FermiNet a démontré pour la première fois comment utiliser l'apprentissage profond pour calculer l'énergie des atomes et des molécules à partir de zéro. Le modèle résultant s'est avéré suffisamment précis pour une application pratique et, au moment de la publication de l'article original (octobre 2020), il restait la méthode de réseau de neurones la plus précise utilisée dans l'industrie. Il est supposéque les méthodes et outils associés peuvent être utiles pour résoudre des problèmes fondamentaux en sciences naturelles. Les auteurs de FermiNet l'utilisent déjà dans leurs travaux sur la convolution des protéines , la dynamique des composés vitreux , la chromodynamique quantique sur un réseau, et dans de nombreux autres projets contribuant à traduire ces développements en pratique.
Une brève histoire de la mécanique quantique
En évoquant la «mécanique quantique», vous risquez de dérouter l'interlocuteur avec ce sujet pas comme les autres. Immédiatement, je me souviens d'images telles que le chat de Schrödinger, qui paradoxalement peut être vivant et mort en même temps, ainsi que des particules élémentaires qui sont à la fois des corpuscules et des ondes. Dans un système quantique, une particule telle qu'un électron n'a pas de localisation spécifique, contrairement à la situation en physique classique. En physique quantique, la position d'un électron est décrite par un nuage de probabilités - c'est-à-dire qu'elle est étalée sur tous ces points, à chacun desquels un électron peut apparaître. En raison de cet état de fait absurde, Richard Feynman a trouvé possible de dire: "Je pense que je peux affirmer avec certitude que personne ne comprend la mécanique quantique."
Malgré toute cette étrange étrangeté, l'essence de la théorie peut être exprimée en quelques équations soignées. La plus célèbre d'entre elles, l'équation de Schrödinger, décrit le comportement des particules à l'échelle quantique de la même manière que les équations de Newton décrivent le comportement des corps aux échelles macroscopiques plus familières. Alors que l'interprétation de cette équation obligera n'importe qui à se saisir de la tête, sa composante mathématique est beaucoup plus facile pour une utilisation pratique, grâce à laquelle le célèbre professeur "tais-toi et compte" est né, avec lequel ils ont combattu les questions philosophiques maladroites des étudiants.
Ces équations suffisent à décrire le comportement de toute matière qui nous est familière au niveau des atomes et des noyaux. Une composante illogique de la mécanique quantique sous-tend toutes sortes de phénomènes exotiques: la supraconductivité, la superfluidité, le laser et les semi-conducteurs ne sont possibles que grâce aux effets quantiques. Mais même une chose aussi modeste qu'une liaison covalente - la composante de base de toute chimie - est le résultat d'interactions quantiques d'électrons. Lorsque ces règles ont finalement été élaborées dans les années 1920, les scientifiques ont réalisé que pour la première fois une théorie avait été créée qui détaillait le travail de toute la chimie. En principe, les équations quantiques pourraient simplement être adaptées pour différentes molécules, résolues en tenant compte de l'énergie du système, puis en déterminant quelles molécules seront stables et quelles réactions se produiront spontanément. Mais,quand on a tenté de s'asseoir et de calculer les solutions de ces équations, il s'est avéré que cela était faisable pour l'atome le plus simple (hydrogène) et pratiquement pas pour aucun autre. Tous les autres calculs se sont avérés trop compliqués.
L'optimisme vertigineux de ces jours a été magnifiquement résumé par Paul Dirac:
Ainsi, les lois physiques de base requises pour une théorie mathématique qui décrirait la plupart de la physique et toute la chimie sont déjà connues. Le hic, c'est qu'en pratique l'application de ces lois donne des équations trop complexes, que nous sommes objectivement incapables de résoudre. Par conséquent, il semble souhaitable de développer des méthodes approximatives pour l'application pratique de la mécanique quantique.
1929
Beaucoup ont repris l'appel de Dirac et bientôt les physiciens ont commencé à développer des méthodes mathématiques qui permettraient d'approcher le comportement des liaisons moléculaires et d'autres phénomènes chimiques à un niveau qualitatif. Tout a commencé par une description approximative du comportement des électrons - cette information est étudiée dans un cours d'introduction à la chimie. Avec cette description, chaque électron est amené dans sa propre orbitale, ce qui vous permet de calculer la probabilité qu'un électron soit trouvé en un point spécifique à proximité d'un noyau atomique. Dans ce cas, la forme de chaque orbitale dépend de la forme moyenne de toutes les autres orbitales. Puisque dans une telle description selon le modèle du «champ auto-cohérent», on suppose que chaque électron est attaché à une seule orbitale, cette image ne transmet que très incomplètement les propriétés réelles des électrons. Pourtant, c'est assezpour déterminer l'énergie totale de la molécule avec une erreur d'environ 0,5% seulement.
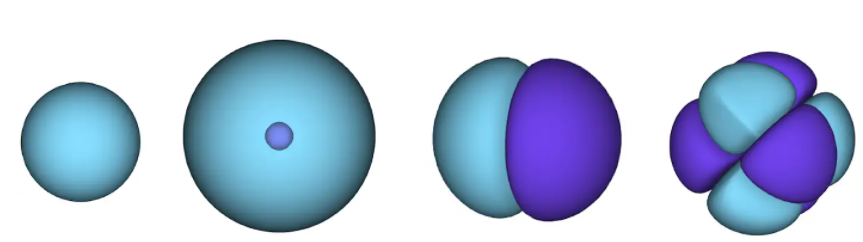
Figure 1 - orbitales atomiques. une surface est une zone dans laquelle un électron est susceptible de se trouver. dans la région bleue, la fonction d'onde est positive et dans le violet, elle est négative.
Malheureusement pour le chimiste en exercice, une erreur de 0,5% est trop importante pour être tolérée. L'énergie des liaisons moléculaires ne représente qu'une petite fraction de l'énergie totale d'un système, et la prédiction correcte de la stabilité d'une molécule peut souvent dépendre d'aussi peu que 0,001% de l'énergie totale du système, soit environ 0,2% de l'énergie de "corrélation" restante.
Par exemple, alors que l'énergie totale des électrons dans une molécule de butadiène est est de près de 100 000 kilocalories par mole, la différence d'énergie entre les différentes configurations possibles de la molécule n'est que de 1 kilocalorie par mole. Autrement dit, s'il est nécessaire de prédire correctement la forme naturelle de la molécule de butadiène, le même niveau de précision est requis que lors de la mesure de la largeur d'un terrain de football avec une précision d'un millimètre.
Avec la prolifération de l'informatique électronique peu après la Seconde Guerre mondiale, les scientifiques ont développé toute une série de méthodes de calcul qui ne pouvaient pas être décrites comme des champs auto-cohérents. Ces méthodes sont désignées par un tas d'abréviations inimaginables couvrant tout l'alphabet, mais chacune de ces méthodes contient une sorte de compromis entre précision et efficacité. À un extrême se trouvent des méthodes qui sont, en fait, précises, mais dont l'échelle est pire qu'exponentiellement à mesure que le nombre d'électrons augmente - elles ne conviennent donc pas pour travailler avec la plupart des molécules, mais les plus petites. À l'autre extrême, il existe des méthodes qui évoluent de manière linéaire mais qui ne sont pas très précises. Ces méthodes de calcul ont eu un impact énorme sur la chimie pratique - le prix Nobel de chimie 1998 a été décerné aux auteurs de plusieurs de ces algorithmes.
Malgré l'étendue des outils de calcul quantique existants, le problème de la représentation efficace de l'information exigeait le développement d'une nouvelle méthode. Ce n'est pas un hasard si seules des dizaines de milliers d'électrons sont impliqués dans les plus grands calculs de chimie quantique modernes (nous parlons des méthodes les plus approximatives), alors que les méthodes classiques de calculs chimiques, par exemple la dynamique moléculaire, permettent de traiter des millions de les atomes. Il n'est pas difficile de décrire l'état d'un système classique - il vous suffit de suivre la position et l'élan de chaque particule. Imaginer l'état d'un système quantique est un défi beaucoup plus grand. Nous devons attribuer une valeur probabiliste à chaque configuration possible de positions d'électrons. Ces informations sont encodées dans une fonction d'onde,vous permettant d'attribuer un nombre positif ou négatif à chaque configuration d'électrons, et la fonction d'onde au carré donne la probabilité avec laquelle le système peut être trouvé dans une telle configuration. L'espace de toutes les configurations possibles est colossal - si vous essayiez de l'imaginer comme une grille avec 100 points dans chaque dimension, alors le nombre de configurations d'électrons possibles pour un atome de silicium serait supérieur au nombre d'atomes dans l'univers!
C'est là que les réseaux de neurones profonds sont utiles. Ces dernières années, d'énormes progrès ont été réalisés dans la représentation de distributions de probabilités complexes à haute dimensionnalité à l'aide de réseaux de neurones. On sait maintenant comment former efficacement de tels réseaux dans l'attente de leur évolutivité. Nous avons suggéré que puisque ces réseaux ont déjà prouvé leur agilité dans la formation de fonctions avec de nombreuses dimensions dans la résolution de problèmes du domaine de l'intelligence artificielle, peut-être qu'ils travailleront pour représenter des fonctions d'ondes quantiques. Nous n'avons pas été les premiers à avoir de telles pensées - d'autres chercheurs, en particulier Giuseppe Carleo et Matthias Troyera démontré comment l'apprentissage profond moderne est applicable à la résolution de problèmes quantiques idéalisés. Nous voulions utiliser des réseaux de neurones pour aborder des problèmes plus réalistes en chimie et en physique du solide, ce qui nous obligeait à prendre en compte les électrons dans nos calculs.
Il n'y a qu'une seule mise en garde lorsque vous travaillez avec des électrons. Les électrons doivent obéir au principe d'exclusion de Pauli, c'est-à-dire que deux électrons ne peuvent pas être simultanément au même endroit. Le fait est que les électrons sont des particules élémentaires parmi les fermions qui composent la majorité des premières briques de matière, en particulier les protons, neutrons, quarks, neutrinos, etc. Leur fonction d'onde doit être antisymétrique - si vous permutez deux électrons, la fonction d'onde est multipliée par -1. Ainsi, il y a une probabilité nulle que deux électrons s'assoient l'un sur l'autre, puisque la probabilité de ceci (et de la fonction d'onde correspondante) est égale à zéro.
Par conséquent, il était nécessaire de développer un réseau de neurones d'un nouveau type, qui serait antisymétrique par rapport à l'entrée qui y pénétrait. Nous l'avons appelé Fermionic Neural Network ou FermiNet. Dans la plupart des méthodes de chimie quantique, l'antisymétrie est introduite à l'aide d'une fonction appelée déterminant. Le déterminant est une matrice qui a la propriété suivante: si vous permutez deux de ses lignes, alors la sortie est multipliée par -1, exactement comme la fonction d'onde des fermions. Vous pouvez prendre un tas de fonctions à un électron, les calculer pour chaque électron de votre système, puis adapter tous les résultats dans une seule matrice. Dans ce cas, le déterminant de la matrice sera une fonction d'onde véritablement antisymétrique. La principale limitation de cette approche est que la fonction résultante - appelée le déterminant de Slater - n'est pas largement applicable.Les fonctions d'onde des systèmes réels sont généralement beaucoup plus complexes. En règle générale, de grandes combinaisons linéaires de déterminants de Slater - parfois des millions ou plus - sont prises pour corriger ce problème, puis quelques corrections simples sont apportées en fonction des paires d'électrons. Même dans ce cas, le système peut ne pas être suffisamment précis pour calculer les énergies.

2 – . – , 1. 1 2 , , -1. .
Les réseaux de neurones profonds sont souvent bien supérieurs en efficacité aux combinaisons linéaires de fonctions de base lors de la représentation de fonctions complexes. Dans FermiNet, cette supériorité est obtenue en introduisant chacune des fonctions dans le déterminant, la fonction de tous les électrons. Cette méthode est beaucoup plus puissante que l'utilisation de fonctions à un et deux électrons. FermiNet fournit un flux d'informations distinct pour chaque électron. Sans tenir compte des interactions entre ces flux, le réseau ne serait pas plus expressif que le déterminant habituel de Slater. Pour faire plus, nous faisons la moyenne des informations collectées à partir de tous les flux sur chacune des couches du réseau et transmettons ces informations à chacun des flux à la couche suivante. En conséquence, ces écoulements ont des propriétés de symétrie appropriées pour créer une fonction antisymétrique.
Les informations sur chacune des couches des réseaux de neurones graphiques sont agrégées de la même manière . Contrairement aux déterminants de Slater, les réseaux FermiNet sont des approximateurs de fonction universels , du moins tant que les couches des réseaux neuronaux restent suffisamment larges. Cela signifie que si nous pouvons former correctement ces réseaux, ils peuvent alors produire une solution presque exacte à l'équation de Schrödinger.
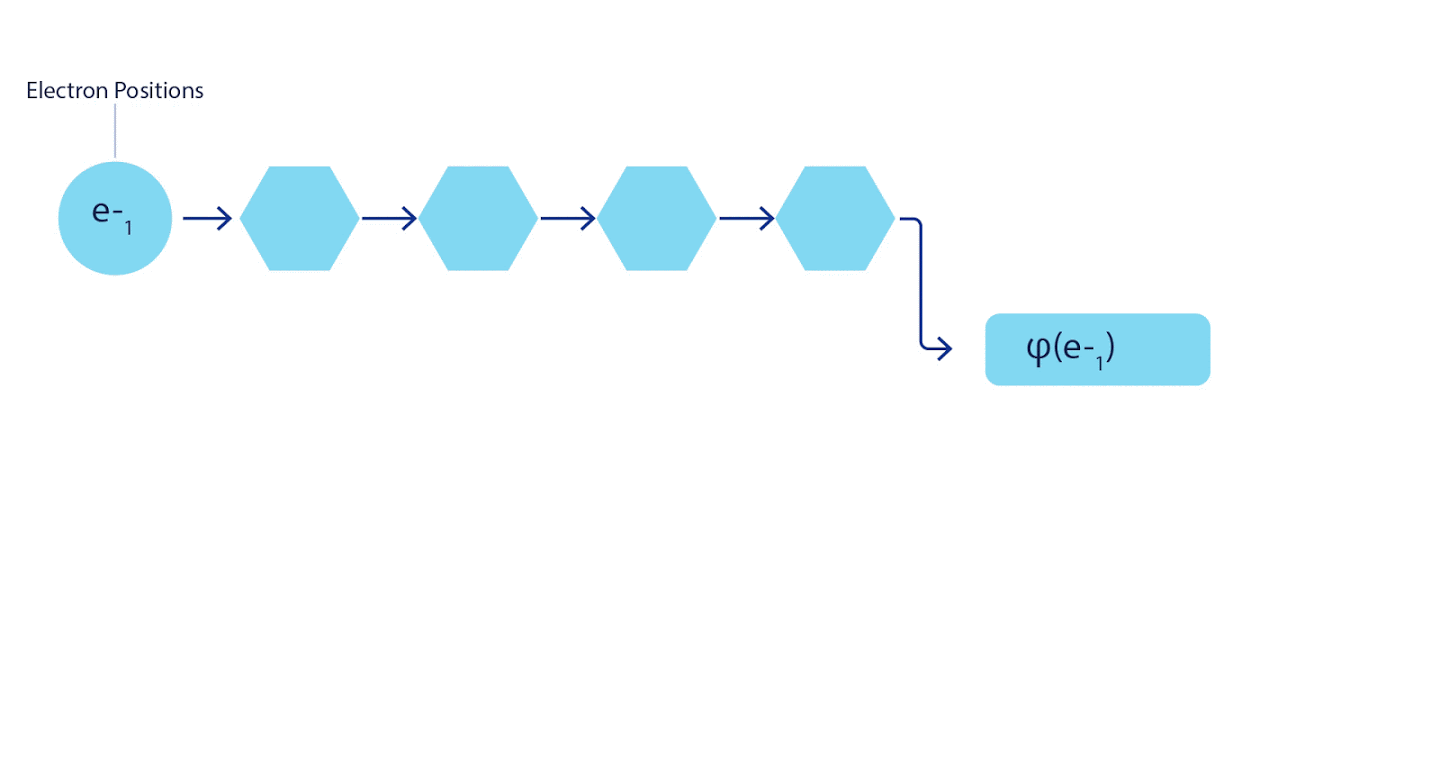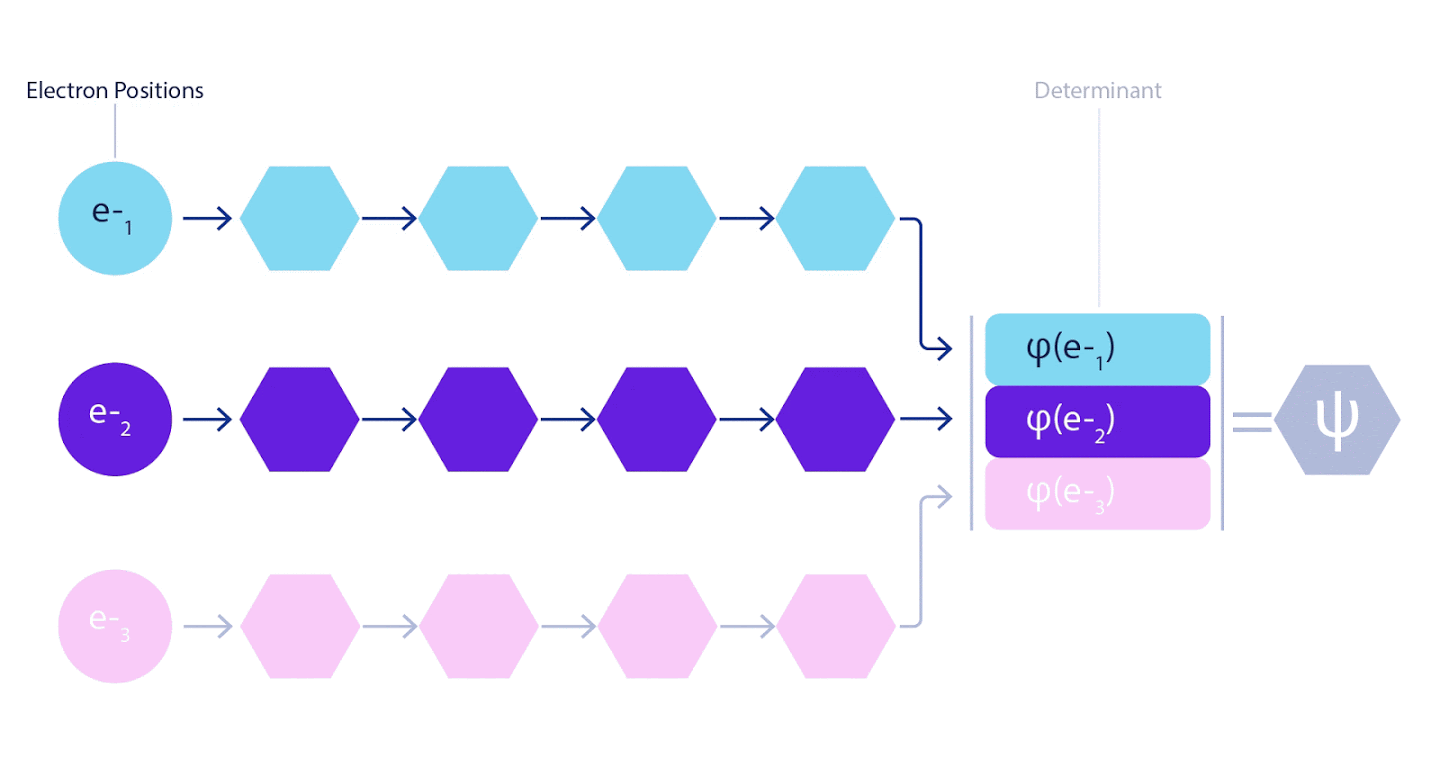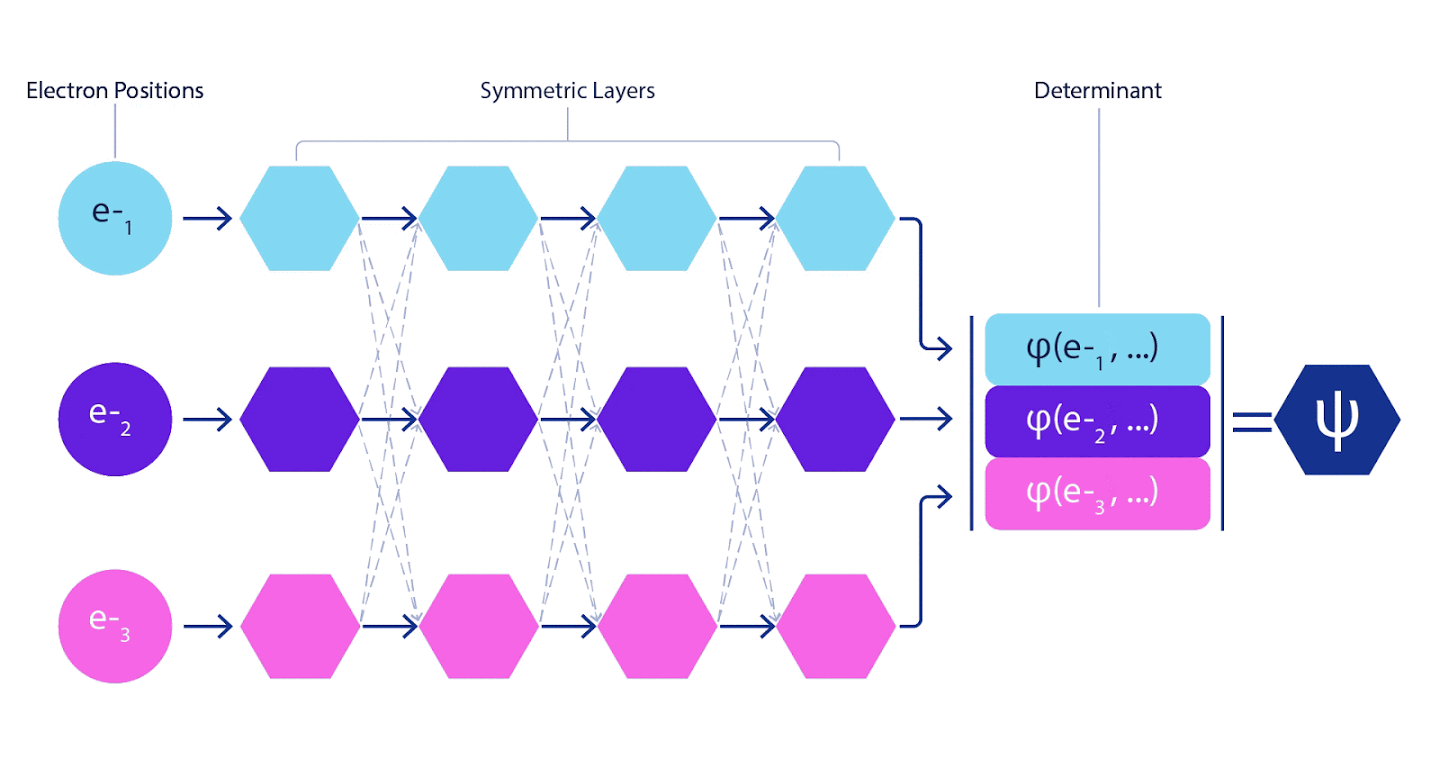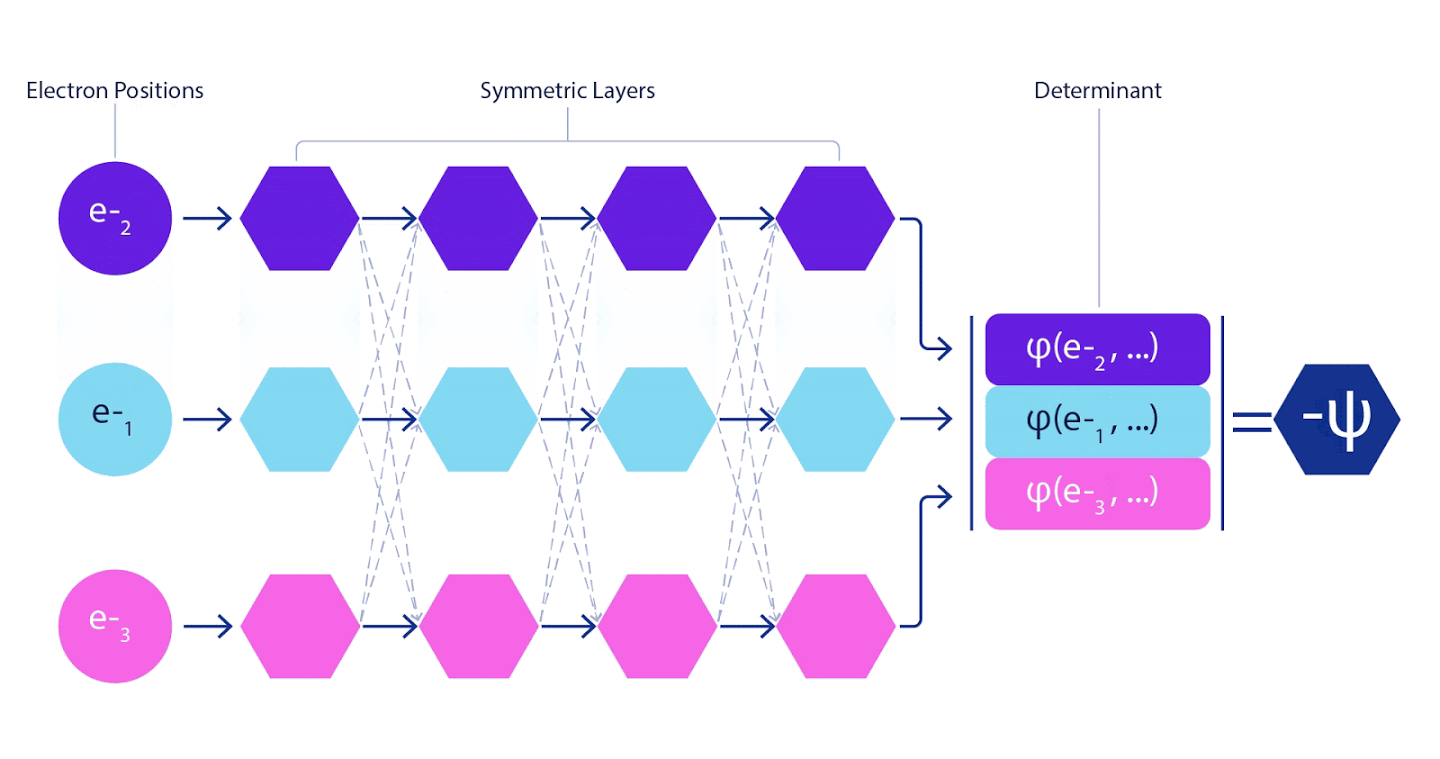
3 – FermiNet. (, ) . FermiNet , , , . , - -1.
Nous personnalisons le réseau FermiNet, minimisant l'énergie du système. Pour faire cela avec précision, nous aurions besoin de calculer la fonction d'onde dans toutes les configurations d'électrons possibles, donc nous devrions le faire approximativement. Par conséquent, nous prenons un échantillon aléatoire de configurations électroniques, calculons l'énergie localement pour chaque variante de l'ordre des électrons et minimisons cette énergie, pas la vraie. Cette méthode s'appelle "Monte Carlo" car c'est un peu comme les actions d'un joueur de casino qui lance les dés encore et encore. Étant donné que la fonction d'onde carrée permet d'observer une configuration de particules à n'importe quel endroit, il est plus pratique de générer des échantillons de la fonction d'onde elle-même - en substance, en simulant l'acte d'observer des particules.
Alors que la plupart des réseaux de neurones sont entraînés sur certaines données externes, dans notre cas, le réseau de neurones lui-même génère l'entrée qui y est utilisée pour la formation. La situation est un peu comme vous sortir d'une tourbière par vos cheveux, et signifie que nous n'avons besoin d'aucune donnée d'entraînement autre que les positions de ces noyaux atomiques autour desquels dansent les électrons. L'idée de base, connue sous le nom de méthode variationnelle quantique de Monte Carlo (ou VMC en abrégé), existe dans la science depuis les années 1960 et est généralement considérée comme un moyen peu coûteux mais pas très précis de calculer l'énergie d'un système. En remplaçant les fonctions d'onde simples basées sur les déterminants de Slater par des fonctions de FermiNet, nous avons réussi à améliorer radicalement la précision de cette approche sur tous les systèmes que nous avons considérés.
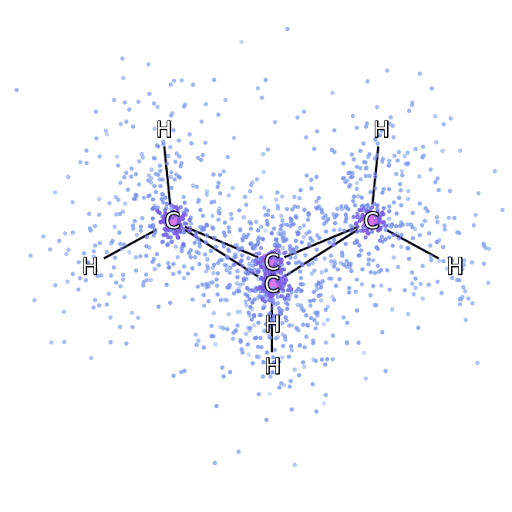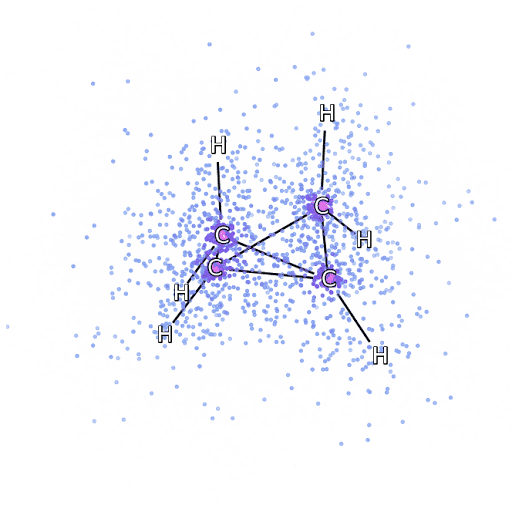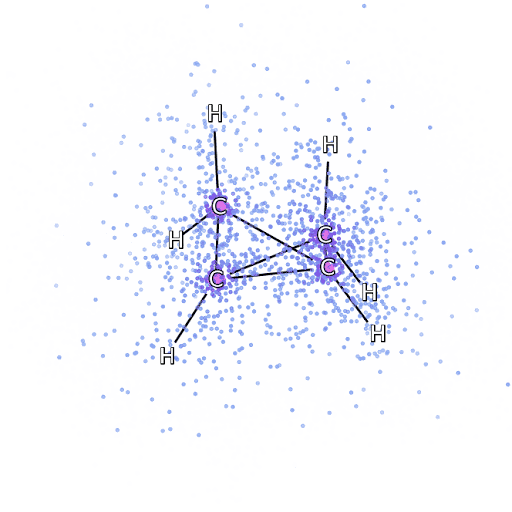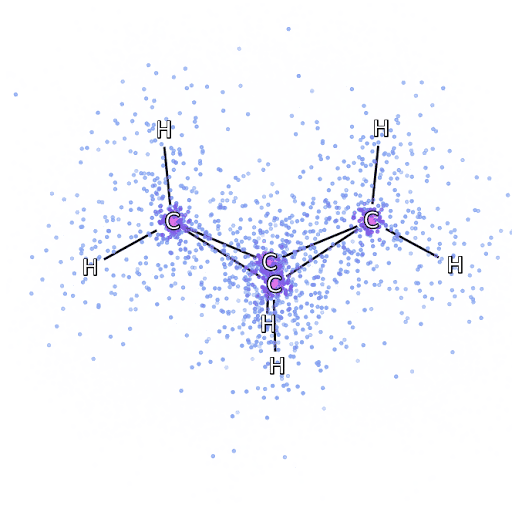
4 – , FermiNet, .
Pour nous assurer que FermiNet est vraiment une percée dans son domaine, nous avons commencé par examiner des systèmes simples et bien étudiés, par exemple, les atomes de la première ligne du tableau périodique (de l'hydrogène au néon). Ce sont de petits systèmes - 10 électrons ou moins - donc ils se prêtent à la recherche en utilisant les méthodes les plus précises (mais exponentiellement plus complexes). FermiNet surpasse de loin les calculs VMC comparables, et peut souvent réduire l'erreur de moitié ou plus par rapport aux calculs exponentiellement évolutifs. Dans les systèmes plus grands, les méthodes qui deviennent plus complexes de manière exponentielle deviennent inapplicables, nous avons donc utilisé la méthode de cluster couplé comme référence. Cette méthode fonctionne bien sur des molécules aux configurations stables, mais glisse lorsque les liaisons sont étirées ou endommagées,et ces facteurs sont essentiels pour comprendre les réactions chimiques. Bien qu'elle évolue beaucoup mieux que de manière exponentielle, la méthode des clusters connectés utilisée dans cette étude fonctionne toujours aussi bien que possible avec des molécules de taille moyenne. Nous avons appliqué FermiNet à des molécules toujours plus grandes, de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons examiné, avec 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».Bien qu'elle évolue beaucoup mieux que de manière exponentielle, la méthode des clusters connectés utilisée dans cette étude fonctionne toujours aussi bien que possible avec des molécules de taille moyenne. Nous avons appliqué FermiNet à des molécules toujours plus grandes, de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons examiné, avec 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».Bien qu'elle évolue beaucoup mieux que de manière exponentielle, la méthode des clusters connectés utilisée dans cette étude fonctionne toujours aussi bien que possible avec des molécules de taille moyenne. Nous avons appliqué FermiNet à des molécules toujours plus grandes, de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons examiné, avec 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».qui a été appliqué dans l'étude décrite, tout le monde travaille également au maximum avec des molécules de taille moyenne. Nous avons appliqué FermiNet à des molécules toujours plus grandes, de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons examiné, avec 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».qui a été appliqué dans l'étude décrite, tout le monde travaille également au maximum avec des molécules de taille moyenne. Nous avons appliqué FermiNet à des molécules toujours plus grandes, de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons examiné, avec 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons envisagé, il a 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».de l'hydrure de lithium au bicyclobutane - c'était le plus grand système que nous ayons envisagé, il a 30 électrons. Sur les plus petites molécules, FermiNet a capturé une étonnante différence de 99,8% entre l'énergie des amas liés et l'énergie dérivée d'un seul déterminant de Slater. Dans le cas du bicyclobutane, FermiNet a quand même capturé 97% ou plus de cette énergie de corrélation - un énorme succès pour une approche soi-disant «bon marché mais imprécise».mais approche imprécise.mais approche imprécise.

La figure 5 est une représentation graphique de la fraction de l'énergie de corrélation que FermiNet capture correctement lors du travail avec des molécules. La barre violette marque 99% d'énergie de corrélation. De gauche à droite: hydrure de lithium, azote, éthylène, ozone, éthanol et bicyclobutane.
Alors que les méthodes de grappes couplées fonctionnent bien avec des molécules stables, la véritable «pointe» de la chimie informatique consiste à comprendre comment les molécules s'étirent, se tordent et se cassent. Lors de la résolution de tels problèmes, les méthodes de cluster connectées échouent souvent, vous devez donc comparer le résultat avec autant d'échantillons de contrôle que possible pour vous assurer que la réponse est cohérente. Dans le cadre de l'expérience décrite, deux systèmes étirés témoins ont été considérés - une molécule d'azote (N 2) et une chaîne hydrogène de 10 atomes (H 10 ). Dans la molécule d'azote, la liaison est particulièrement complexe, puisque 3 électrons de chaque atome y participent.
La chaîne de l'hydrogène, quant à elle, est intéressante pour comprendre les propriétés des électrons dans les matériaux , par exemple pour prédire si un matériau donné conduira l'électricité ou non. Dans les deux systèmes, la méthode des clusters connectés fonctionnait bien en équilibre, mais se heurtait à des difficultés lorsque les liaisons étaient étirées. Les méthodes VMC traditionnelles n'ont pas bien fonctionné dans l'ensemble de la gamme d'exemples. Mais FermiNet s'est avéré être l'une des meilleures méthodes de toutes les recherches, quelle que soit la longueur de la liaison.
Conclusion
Nous pensons que FermiNet est le début de grandes avancées dans la synthèse des méthodes d'apprentissage en profondeur et de la chimie quantique computationnelle. La plupart des systèmes avec lesquels FermiNet a été examiné jusqu'à présent sont bien compris et compris. Mais tout comme les premiers bons résultats utilisant l'apprentissage profond dans d'autres domaines ont stimulé une vague de recherches supplémentaires et des progrès rapides, nous espérons que la même chose se produira avec FermiNet, et des idées pour de nouvelles architectures de réseau neuronal encore meilleures émergeront. Déjà après la publication du travail décrit sur arXiv, d' autres groupesont partagé leurs approches d'application de l'apprentissage en profondeur pour résoudre des problèmes impliquant de nombreux électrons. De plus, nous venons à peine de creuser dans la physique quantique computationnelle et prévoyons d'utiliser FermiNet pour résoudre des problèmes complexes dans le domaine de la science des matériaux et de la physique du solide.
L'article scientifique est ici , et le code peut être consulté ici . Les auteurs tiennent à remercier Jim Kinwin, Adam Kine et Dominic Barlow pour leur aide dans la préparation des dessins.