
Les mathématiques sont souvent appelées le «langage de la science». Il convient parfaitement au traitement quantitatif de presque toutes les informations scientifiques, quel que soit leur contenu. Et avec l'aide du formalisme mathématique, les scientifiques de différents domaines peuvent, dans une certaine mesure, se «comprendre». Aujourd'hui, une situation similaire émerge avec l'informatique. Mais si les mathématiques sont le langage de la science, alors CS est son couteau suisse. En effet, il est difficile d'imaginer la recherche moderne sans analyser et traiter d'énormes quantités de données, des calculs complexes, la modélisation informatique, la visualisation et l'utilisation de logiciels et d'algorithmes spéciaux. Regardons quelques «histoires» intéressantes lorsque différentes disciplines utilisent des méthodes CS pour résoudre leurs problèmes.
Bioinformatique: de la boîte de Pétri à la biologie In silico
La bioinformatique peut être considérée comme l'un des exemples les plus frappants de l'intersection de la CS et d'autres disciplines. Cette science traite de l'analyse de données biologiques moléculaires à l'aide de méthodes informatiques. La bioinformatique en tant que direction scientifique distincte est apparue au début des années 70 du siècle dernier, lorsque les séquences nucléotidiques de petits ARN ont été publiées pour la première fois et que des algorithmes de prédiction de leur structure secondaire (la disposition spatiale des atomes dans une molécule) ont été créés.
Une nouvelle ère de la bioinformatique a commencé avec le Human Genome Project, qui vise à déterminer la séquence des nucléotides dans l'ADN humain et à identifier les gènes du génome. Le coût du séquençage de l'ADN (séquençage des nucléotides) a baissé de plusieurs ordres de grandeur. Cela a conduit à une augmentation considérable du nombre de séquences dans les bases de données publiques. Le graphique ci-dessous montre la croissance du nombre de séquences dans la base de données publique GenBank de décembre 1982 à février 2017 sur une échelle semi-logarithmique. Pour que les données accumulées deviennent utiles, elles doivent être analysées d'une manière ou d'une autre.

Croissance du nombre de séquences dans GenBank de décembre 1982 à février 2017. Source: www.ncbi.nlm.nih.gov/genbank/statistics
L'une des méthodes d'analyse de séquence en bioinformatique est l'alignement de séquence. L'essence de la méthode réside dans le fait que les séquences de monomères d'ADN, d'ARN ou de protéines sont placées les unes sous les autres de manière à voir des zones similaires. La similitude des structures primaires (c'est-à-dire des séquences) de deux molécules peut refléter leur relation fonctionnelle, structurelle ou évolutive. Puisqu'une séquence peut être représentée comme une chaîne avec un alphabet spécifique (4 nucléotides pour l'ADN et 20 acides aminés pour la protéine), l'alignement s'avère être une tâche combinatoire de CS (par exemple, l'alignement de ligne est également utilisé dans le traitement du langage naturel - PNL). Cependant, le contexte de la biologie ajoute une certaine spécificité au problème.
Regardons l'alignement en utilisant des protéines à titre d'exemple. Un résidu d'acide aminé dans la protéine correspond à une lettre de l'alphabet latin dans la séquence. Les chaînes sont écrites l'une en dessous de l'autre pour obtenir la meilleure correspondance. Les éléments correspondants sont les uns en dessous des autres, les "espaces" sont remplacés par "-" (espace). Ils désignent indel , c'est-à-dire le lieu d'insertion possible (introduction dans une molécule d'un ou plusieurs nucléotides ou acides aminés) et des délétions («décrochage» d'un nucléotide ou d'un acide aminé).

Un exemple de l'alignement des séquences d'acides aminés de deux protéines. La leucine (L) et l'isoleucine (I), qui sont des isomères, sont surlignées en bleu - une telle substitution dans la plupart des cas n'affecte pas la structure protéique
Cependant, comment pouvez-vous déterminer si l'alignement est optimal? La première chose qui me vient à l'esprit est d'estimer le nombre de matchs: plus il y a de matchs, mieux c'est. Cependant, dans le contexte de la biologie, ce n'est pas tout à fait vrai. Les substitutions (substitutions d'un acide aminé par un autre) sont inégales: certaines substitutions (par exemple, S et T, D et E sont des résidus dont la structure diffère d'exactement un atome de carbone) n'affectent pratiquement pas la structure des protéines. Mais le remplacement de la sérine par du tryptophane modifiera considérablement la structure de la molécule. Un critère quantitatif (poids ou score) est saisi pour déterminer si la compensation est la meilleure possible. Pour évaluer les substitutions, on utilise des matrices dites de substitution, basées sur les statistiques de substitution d'acides aminés dans des protéines de structure connue. Plus le nombre à l'intersection des lettres correspondantes est élevé, plus le score est élevé.

De nouvelles matrices de substitution apparaissent périodiquement. Voici la matrice BLOSUM 62.
Le score prend également en compte la présence de suppressions. Habituellement, la pénalité pour «ouvrir» une suppression est de plusieurs ordres de grandeur plus élevée que pour «continuer». Cela est dû au fait qu'une section de plusieurs lacunes consécutives est considérée comme une mutation, et plusieurs lacunes à différents endroits sont considérées comme plusieurs. Dans l'exemple ci-dessous, la première paire de séquences est plus similaire que la seconde, car dans le premier cas, les séquences sont formellement séparées par un événement évolutif:

Maintenant sur les algorithmes d'alignement eux-mêmes. Il existe deux types d'alignement apparié (recherche de zones similaires de deux séquences): global et local. L'alignement global implique que les séquences sont homologues (similaires) sur toute leur longueur. Il comprend les deux séquences dans leur intégralité. Cependant, avec cette approche, des domaines similaires ne sont pas toujours bien définis s'ils sont peu nombreux. L'alignement local est utilisé si les séquences sont conservées comme homologues (par exemple, en raison d'une recombinaison) et sites indépendants. Mais il ne peut pas toujours entrer dans la zone d'intérêt, de plus, il y a une possibilité de rencontrer une zone similaire accidentelle. Pour obtenir un alignement par paires, des méthodes de programmation dynamique sont utilisées (résoudre un problème en le divisant en plusieurs sous-tâches identiques connectées de manière récurrente). Dans les programmes d'alignement global, l'algorithme Needleman-Wunsch est souvent utilisé , et pour l' alignement local, l'algorithme Smith-Waterman . Vous pouvez en savoir plus à leur sujet en suivant les liens.
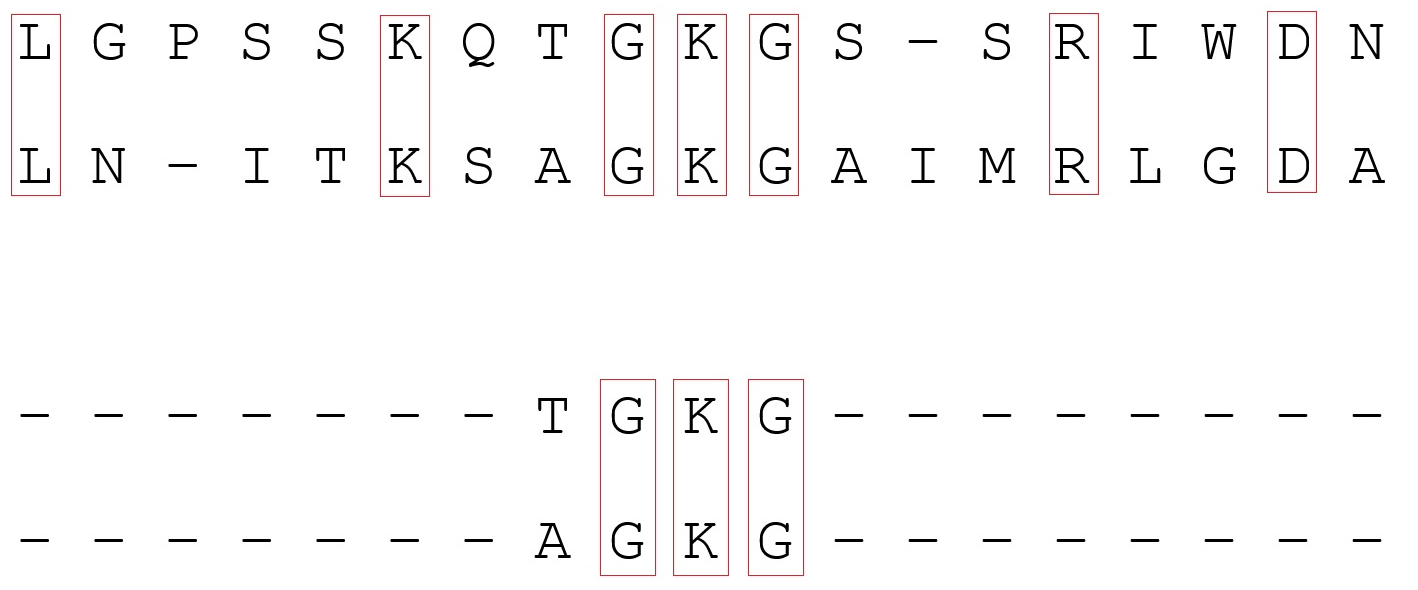
Exemple d'alignement: le haut est global, le bas est local. Dans le premier cas, l'alignement se produit sur toute la longueur des séquences; dans le second, certaines régions homologues sont trouvées.
Comme vous pouvez le voir, la tâche biologique peut être tout à fait réduite à la tâche de CS. L'alignement par paires utilisant les algorithmes mentionnés nécessite environ m * n de mémoire supplémentaire (m, n sont les longueurs des séquences), que les ordinateurs domestiques modernes peuvent facilement gérer. Cependant, la bioinformatique a également des tâches plus non triviales, par exemple, l'alignement multiple (alignement de plusieurs séquences) pour la reconstruction d' arbres phylogénétiques... Même si nous comparons 10 très petites protéines avec une longueur de séquence d'environ 100 caractères, alors une quantité inacceptable de mémoire supplémentaire sera nécessaire (la dimension du tableau est 100 ^ 10). Par conséquent, dans ce cas, l'alignement est basé sur diverses heuristiques.
Modélisation de la structure à grande échelle de l'univers
Contrairement à la biologie, la physique côtoie l'informatique depuis les débuts de l'informatique. Avant la création des premiers ordinateurs, le mot «ordinateur» (calculatrice) était appelé une position spéciale - il s'agissait de personnes qui effectuaient des calculs mathématiques sur des calculatrices. Ainsi, lors du projet Manhattan, le physicien Richard Feynman était le directeur de toute une équipe de "calculateurs" qui traitaient les équations différentielles sur les machines à additionner.

"Salle de calcul" du Flight Research Center. Armstrong. USA, 1949
À l'heure actuelle, les méthodes CS sont largement utilisées dans divers domaines de la physique. Par exemple, la physique computationnelle étudie des algorithmes numériques pour résoudre des problèmes physiques pour lesquels une théorie quantitative a déjà été développée. Dans les situations où l'observation directe des objets est difficile (cela se produit souvent en astronomie), la modélisation informatique vient en aide aux scientifiques. Un tel cas est précisément l'étude de la structure à grande échelle de l'Univers : les observations d'objets distants sont difficiles en raison de l' absorption du rayonnement électromagnétique dans le plan de la Voie lactée, la modélisation est donc devenue la principale méthode de recherche.

,
L'une des tâches de la cosmologie moderne est d'expliquer l'image observée de la diversité des galaxies et de leur évolution. Au niveau qualitatif, les processus physiques se produisant dans les galaxies sont maintenant connus, par conséquent, les efforts des scientifiques visent à obtenir des prédictions quantitatives. Cela permettra de répondre à un certain nombre de questions fondamentales, par exemple sur les propriétés de la matière noire. Mais, avant d'isoler les manifestations observées de la matière noire, il est nécessaire de comprendre le comportement de la matière ordinaire. À grande échelle (plusieurs millions d'années-lumière), la matière ordinaire se comporte effectivement de la même manière que l'obscurité: elle est soumise à une force de gravité, vous pouvez oublier la pression du gaz. Cela permet de simuler relativement facilement l'évolution de la structure à grande échelle de l'Univers (méthodes numériques,contenant uniquement de la matière noire ou semblable à de la poussière et reproduisant bien la structure à grande échelle de la distribution des galaxies, a commencé à se développer depuis les années 1980)
La matière noire est modélisée comme suit. Le cube virtuel, d'une taille de centaines de millions d'années-lumière, est presque uniformément rempli de particules d'essai - des corps. Dès le début, de petites inhomogénéités étaient présentes dans l'Univers, d'où toute la structure observée est née, par conséquent, le remplissage est "presque uniforme". Puis les particules commencent à «vivre leur propre vie» sous l'influence de la gravité: le problème des N corps est résolu . Les particules échappées du cube sont transférées sur la face opposée et les forces gravitationnelles se propagent également avec le transfert. Grâce à cela, le cube devient pour ainsi dire infini, comme l'univers.

Trajectoires approximatives de trois corps identiques situés aux sommets d'un triangle non isocèle et ayant des vitesses initiales nulles
L'un des modèles numériques les plus connus de ce type est le Millenium , qui a une taille de cube de plus de 1,5 milliard d'années-lumière et environ 10 milliards de particules. Dans les années suivantes, plusieurs modèles plus grands ont été réalisés: l' Horizon Run avec un côté cube 4 fois plus grand que le Millenium, et le Dark Sky avec 16 fois le Millenium. Ces modèles et des modèles similaires ont joué un rôle clé dans les projets de validation du modèle Lambda-CDM désormais généralement accepté. (Un univers contenant environ 70% d'énergie noire, 25% de matière noire et 5% de matière ordinaire).

Millenium: , ; — . .
La réduction d'échelle pose des problèmes de mise en correspondance des observations et des modèles numériques avec une seule matière noire. À plus petite échelle (l'échelle de propagation des ondes de choc à partir des supernovae), la matière ne peut plus être considérée comme poussiéreuse. Il est nécessaire de prendre en compte l'hydrodynamique, le refroidissement et le chauffage du gaz par rayonnement, et bien plus encore. Pour prendre en compte toutes les lois de la physique dans la modélisation, quelques simplifications sont apportées: par exemple, vous pouvez diviser le cube modèle en un réseau de cellules (physique du sous-réseau), et supposer que lorsqu'une certaine densité et température dans la cellule est atteinte , une partie du gaz se transforme instantanément en étoile. Cette classe de modèles comprend les projets EAGLE et illustris . L'un des résultats de ces projets est la reproduction de la relation Tully-Fisher entre la luminosité de la galaxie et la vitesse de rotation du disque.
Linguistique et apprentissage automatique: un pas de plus vers la résolution d'un mystère vieux de 4000 ans
Les méthodes CS trouvent des applications dans des domaines plus inattendus, par exemple dans l'étude des langues anciennes et des systèmes d'écriture. Ainsi, une étude d'un groupe de scientifiques dirigé par Rajesh P.N. Rao, professeur à l'Université de Washington, a mis en lumière le mystère de l'écriture de la vallée de l'Indus.
L'écriture Indus, utilisée entre 2600-1900 avant JC dans ce qui est aujourd'hui le Pakistan oriental et le nord-ouest de l'Inde, appartenait à une civilisation non moins complexe et mystérieuse que ses contemporains mésopotamiens et égyptiens. Il en reste très peu de sources écrites: les archéologues n'ont trouvé qu'environ 1 500 inscriptions uniques sur des fragments de céramique, des tablettes et des sceaux. Le lettrage le plus long ne comporte que 27 caractères.

Inscriptions sur les phoques de la vallée de l'Indus
Dans la communauté scientifique, il y avait diverses hypothèses sur les «symboles mystérieux». Certains experts considéraient que les symboles n'étaient rien de plus que de «jolies images». Ainsi, en 2004, le linguiste Steve Farmer a publié un article dans lequel il a été soutenu que l'écriture de l'Indus n'est rien de plus que des symboles politiques et religieux. Sa version, bien que controversée, trouve toujours ses partisans.
Rajesha P.N. Rao, un scientifique en apprentissage automatique, a lu des articles sur l'écriture Indus au lycée. Un groupe de scientifiques sous sa direction a décidé de mener une analyse statistique des documents fiables existants. Au cours de recherches utilisant des chaînes de Markov(l'une des premières disciplines dans laquelle les chaînes de Markov ont trouvé une application pratique était la critique textuelle) l' entropie conditionnelle a été comparée symboles de l'écriture Indus avec l'entropie des séquences de signes linguistiques et non linguistiques. L'entropie conditionnelle est l'entropie d'un alphabet pour lequel les probabilités d'une lettre après l'autre sont connues. Plusieurs systèmes ont été sélectionnés pour comparaison. Les systèmes linguistiques comprenaient: l'écriture logographique sumérienne, le vieux tamoul Abugida, le sanscrit du Rig Veda, l'anglais moderne (les mots et les lettres ont été étudiés séparément) et le langage de programmation Fortran. Les systèmes non linguistiques ont été divisés en deux groupes. Le premier comprenait des systèmes avec un ordre rigide de signes (ensemble artificiel de signes n ° 1), le second - des systèmes avec un ordre flexible (protéines de bactéries, ADN humain, ensemble artificiel de signes n ° 2). En conséquence, il s'est avéré que l'écriture proto-indienne s'est avérée modérément ordonnée, comme l'écriture des langues parlées:l'entropie des documents existants est similaire à l'entropie des scripts sumérien et tamoul.

Entropie conditionnelle pour divers systèmes linguistiques et non linguistiques
Ce résultat réfute l'hypothèse de l'usage ornemental des signes. Et bien que les méthodes CS aient aidé à confirmer la version selon laquelle les symboles de la vallée de l'Indus sont très probablement un système d'écriture, la question n'est pas encore arrivée au décryptage.
Conclusion
Bien sûr, de nombreux domaines dans lesquels les méthodes CS trouvent une application sont exagérés. Il est tout simplement impossible dans un article de révéler comment la science moderne repose sur la technologie informatique. Cependant, j'espère que les exemples donnés montrent comment différents problèmes peuvent être résolus, y compris par les méthodes CS.
Les serveurs cloud de Macleod sont rapides et sécurisés.
Inscrivez-vous en utilisant le lien ci-dessus ou en cliquant sur la bannière et bénéficiez d'une remise de 10% pour le premier mois de location d'un serveur de toute configuration!
